As a medical student I was taught the CKD hypertension gospel straight from the good book of
JNC VII: Thou shalt lower the blood pressure to less than 130/80! This was many years after David Bowe and Freddie Mercury but I got the song stuck in my head when I started thinking about the post so I had to put it up there.
I lived happily with this for many years until one day someone questioned me. Why should you lower the blood pressure to less than 130/80 in someone with chronic kidney disease? Well 'cause the JNC VII says so! Check it out...

Right there in red, blue and black. And supported by two references no less! One of them is the American Diabetic Association going on about diabetes (another story) but reference 21 is
KDOQI on CKD... So the rabbit hole gets deeper.
Over at KDOQI we get the following...

They say "controlled trials in essential hypertension conclusively show a beneficial effect of lowering blood pressure to <140/90 mm Hg. Controlled trials in high-risk individuals with diabetes or heart failure suggest beneficial effects of reduction of blood pressure to even lower values. Based on these studies, and on observational studies, a number of guidelines for patients with either diabetes mellitus or congestive heart failure recommend a goal blood pressure of <130/80 mm Hg.
There are few studies regarding blood pressure goals for CVD risk reduction in patients with CKD. Thus, the Work Group elected to extrapolate the recommendations for high-risk patients to patients with CKD."
Uhh so, we have no evidence so we took some evidence from other diseases and said do the same thing. It not quite that bad. There is some evidence for less than 130/80 but it has caveats.
The
MDRD study randomized patients to aggressive vs standard blood pressure control with achieved average values of 126/77 and 133/80 respectively. At the end of the study there was no overall difference between the two groups in terms of kidney function but in post-hoc analysis the aggressive BP arm had statistically slower rates of renal function decline in patients with over 1g per day of proteinuria mainly driven by patients with over 3g of proteinuria per day. Unfortunately, the aggressive control group were more likely to have received ACE inhibitors than the standard control group so the post-hoc data is a bit muddled.
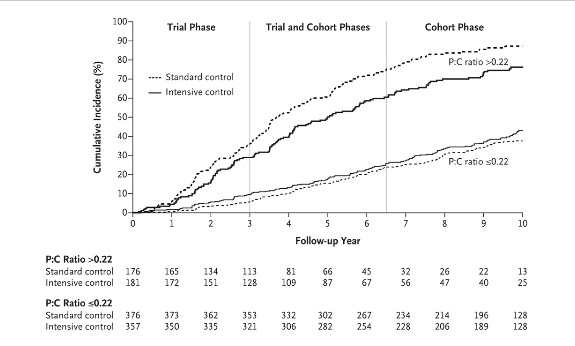
In the recently published long term followup of the
AASK trial, African Americans with hypertensive kidney disease who were initially randomized to either intensive or standard BP control were subsequently followed in a cohort phase in which the BP target was the same in both groups. Followup extended out to 12 years from the initial randomization. The achieved BPs during the trial were 130/78 mm Hg vs 141/86 mm in the intensive and standard groups respectively. In the cohort phase BPs were much closer as expected (131/78 and 134/78 in the intensive and standard groups respectively).
The story is similar to MDRD, among all patients there was no difference in the primary composite outcome of ESRD, doubling of serum creatinine or death throughout the trial and cohort phase. However, in the subgroup with baseline proteinuria of greater than 220 mg per day a significant difference between BP target groups appeared favoring more intensive control.

So no clean randomized prospective data to support the JNC VII target of less than 130/80 in CKD patients. There is a hint from the above subgroup analyses that CKD patients with proteinuria might benefit from having blood pressures controlled to below 130/80. The proteinuria cutpoint at which this might occur is unclear.
It will be interesting to see how JNC VIII, expected sometime later this year, handles the above. Additional information will hopefully come from the randomized prospective
SPRINT trial which is looking at systolic BP goals of 140 vs 120 in a large cohort with a reasonable proportion of CKD patients.